“Questo risultato riveste un grande valore scientifico”- commenta il Prof. Generoso Andria, Primario dell’Unità Operativa Complessa di Genetica Clinica Pediatrica dell’Azienda Ospedaliero-Universitaria Federico II – ” come testimoniato anche dal grande prestigio della rivista scientifica sulla quale è stata pubblicata la ricerca, ovvero Nature Medicine”.
“Lo studio – continua il Prof. Andria – dimostra che il difetto anche parziale della proteina nexina 27 causa nel modello animale alterazioni della funzione delle cellule neuronali con disturbi della memoria e dell’apprendimento.
L’elemento interessante che collega la scoperta alla sindrome di Down è che nelle cellule con trisomia del cromosoma 21 esiste un’espressione aumentata del microRNA 155 (una molecola importante per la regolazione dei geni, presente proprio sul cromosoma 21) che contribuisce a ridurre i livelli di nexina 27 nei modelli sperimentali di sindrome di Down”. Il prof. Andria sottolinea infine che è ancor più interessante l’osservazione che, aumentando i livelli di nexina 27 nel cervello di modelli animali di sindrome di Down con metodi di biologia molecolare, si osserva il miglioramento sia della funzione dei neuroni che dell’apprendimento.
Questa scoperta di ricerca di base definisce quindi un nuovo meccanismo molecolare che può contribuire alla patogenesi della sindrome di Down e offre la base per studi di modulazione della proteina in modelli sperimentali, con la speranza di poter traslare i risultati della ricerca nell’uomo.
Queste proprietà hanno subito interessato due ricercatrici del CNR di Bari – le dott.sse Rosa Vacca e Daniela Valenti che, in collaborazione con il Dipartimento di Pediatria dell’Università Federico II di Napoli e i Dipartimenti di Scienze Mediche di Base, Neuroscienze e Organi di Senso dell’Università di Bari, di Clinica e Medicina Sperimentale dell’Università di Pisa, hanno dimostrato che nella sindrome di Down aumenta lo stress ossidativo ed è fortemente compromessa la funzionalità mitocondriale.
L’alterata funzionalità mitocondriale nei pazienti sembra essere una delle cause determinanti del deficit intellettivo e della neuro-degenerazione precoce.
Trattando con EGCG cellule della pelle (fibroblasti) e del sangue (linfoblastoidi, derivati dei linfociti) lo studio ha riscontrato una riattivazione funzionale dei complessi respiratori mitocondriali, un incremento della produzione da parte dei mitocondri di adenosina trifosfato (Atp), cioè la principale fonte di energia cellulare, una diminuzione dei livelli di specie reattive dell’ossigeno (Ros) e un aumento del numero dei mitocondri.
L’EGCG è nota per le sue proprietà antitumorali e antinfiammatorie ed è già testata sull’uomo.
Questi risultati propongono quindi l’EGCG come possibile candidato per il trattamento di questa patologia, sperimentazione che passerà prima dal modello animale per poi passare alla fase clinica.
I ricercatori hanno evidenziato che cellule cutanee provenienti da persone Down (fibroblasti) sono più suscettibili all’apoptosi, ovvero alla morte cellulare in seguito a specifici stimoli, e che questo potrebbe dipendere dal potenziale della membrana mitocondriale e dalla membrana mitocondriale esterna.
E’ ormai noto che la carenza di acido folico nelle donne al momento del concepimento può aumentare il rischio di alcune malformazioni tra le quali la spina bifida ed alcuni difetti cardiaci. Proprio in virtù di ciò, da qualche anno si raccomanda l’assunzione di un supplemento di acido folico a tutte le donne in età fertile che programmano una gravidanza o che non ne escludano attivamente la possibilità. E infatti, in Italia sono prescrivibili dai medici di famiglia formulazioni di acido folico al dosaggio di 0,4 mg a carico del sistema sanitario nazionale per la riduzione del rischio di queste malformazioni.
L’acido folico, ed i geni che servono al suo metabolismo, sono stati anche studiati nelle mamme di bambini con sindrome di Down.
Già nel 2006, presso il Dipartimento di Pediatria dell’Università di Napoli Federico II, il Prof. Generoso Andria e la Dott.ssa Iris Scala hanno condotto uno studio sulla frequenza di alcune varianti di geni del metabolismo dell’acido folico, dimostrando che alcune varianti geniche, da sole o tra loro associate, erano più frequenti nelle donne che avevano avuto un bambino con la sindrome di Down rispetto a donne di controllo.
I risultati di questo studio, pubblicato su una nota rivista internazionale, Genetics in Medicine, sono stati ripresi da un recente studio, pubblicato nei primi mesi del 2013, che ha confermato l’associazione tra una variante genica chiamata MTHFR C677T e un modico aumento del rischio di concepire un bambino con la sindrome di Down nelle donne portatrici.
Questo significa che tutte le donne dovrebbero fare l’analisi genetica? No. Allo stato attuale non ci sono indicazioni abbastanza forti per estendere l’analisi genetica a tutta la popolazione. I risultati di questa ricerca incoraggiano tuttavia a porre attenzione alla corretta alimentazione in età fertile favorendo un alto apporto di acido folico, eventualmente supplementato con i preparati presenti in commercio.
Dr.ssa Iris Scala
La gioia e il dolore, la paura e l’impazienza si mescolavano in un vortice di sentimenti che avrebbe avuto il suo culmine quando finalmente, esausta, sentì il primo vagito. Non sapeva ancora il sesso del suo bambino, “preferiamo la sorpresa!”, e allora che sorpresa sia! Le parole del medico si sarebbero stampate per sempre nella sua memoria, lo sapeva, ma non furono quelle che si aspettava: “Il suo bambino ha la sindrome di Down…”
E’ così che comincia la storia di un bambino su 1000, una storia che sarà di ragazzi, di donne e di uomini che, a causa di un solo, piccolo cromosoma in più, dovranno affrontare più difficoltà e incomprensioni, e per i quali tutto costerà più fatica e sforzo rispetto a tutti quanti gli altri. Ed è la storia che ognuno di noi si augura gli venga raccontata da qualcun altro, sebbene chi incontra questa sindrome sulla propria strada presto si accorge di trovarsi a contatto con persone di incredibile dolcezza, sensibilità e capacità di donare affetto. Ebbene sì, la sindrome di Down è anche questo: innocenza, candore, emotività e tenerezza disarmanti.
Perché allora la vita di una persona con la sindrome di Down è ancora così difficile? Non è stato forse fatto abbastanza? Vediamo.
Molte delle manifestazioni cliniche della sindrome di Down, tra cui potremmo citare disturbi cardiovascolari, ipotiroidismo, diabete, celiachia e cataratta, possono essere tenute sotto controllo, il che ha significativamente migliorato l’aspettativa e la qualità della vita. Ciò ha implicato progressi notevoli anche da un punto di vista sociale: le persone con sindrome di Down vengono oggi inserite in corsi di studio regolari, e non più nelle cosiddette “scuole di sostegno”, e in contesti lavorativi in cui ricevono incarichi che possano svolgere in piena autonomia.
D’altro canto, però, l’allungamento della vita media delle persone con sindrome di Down ha comportato l’insorgenza di ulteriori sintomi clinici, quali cataratta, malattia di Alzheimer, diabete mellito e artrite reumatoide che, pur essendo tipici dell’età senile nella popolazione normale, compaiono precocemente nelle persone con sindrome di Down. Il quadro clinico di una persona adulta è quindi piuttosto complesso e richiede un monitoraggio costante e approcci terapeutici mirati a controllare a posteriori le singole manifestazioni cliniche. Sarebbe invece auspicabile un tipo di intervento terapeutico causale, col quale poter prevenire almeno le patologie ad essa correlate.
Un tale traguardo richiede ancora molti studi e ricerche, dal momento che sono ancora troppe le cose che non sappiamo, prima fra tutte il rapporto causa-effetto tra i geni del cromosoma 21, presenti in tre copie, e le manifestazioni cliniche; è questa la chiave essenziale per comprendere le basi molecolari responsabili di un quadro clinico così complesso. Molto è stato fatto, non c’è dubbio, e lo studio di modelli animali della trisomia sempre più sofisticati, insieme con la conoscenza dettagliata di tutta la mappa del cromosoma 21 e dei geni che lo costituiscono, ha contribuito in maniera sostanziale a chiarire molti dei troppi aspetti ancora oscuri di questa complessa sindrome.
Uno di questi geni, chiamato CBS, ha un ruolo fondamentale nel metabolismo degli amminoacidi solforati ed è stato il punto di partenza di un recente studio che ha coinvolto il gruppo del Prof. Vincenzo Zappia, del Dipartimento di Biochimica e Biofisica della Seconda Università degli Studi di Napoli, in collaborazione col Dipartimento di Pediatria dell’Università di Napoli “Federico II”, diretto dal Prof. Generoso Andria in accordo con l’Associazione Sindrome di Down. Da tale studio è emerso che le proteine di membrana del globulo rosso di bambini con sindrome di Down presentano un’alterazione chimica che potrebbe avere conseguenze sulla struttura e la funzione delle proteine stesse e, indirettamente, sulla funzione dell’intera cellula. Tale alterazione si verifica, con molta probabilità, in risposta all’esposizione cronica a radicali liberi, cui tali proteine vanno incontro nel corso del tempo, in misura maggiore negli individui con sindrome di Down.
E’ ancora presto per valutare l’impatto di questa alterazione sul quadro clinico della sindrome, ma ci si può augurare sin d’ora che un protocollo terapeutico che preveda l’uso di particolari molecole potrà sicuramente essere di aiuto. Questo è però soltanto uno degli aspetti del complesso mondo Down, e la strada da percorrere è ancora molto lunga.
La ricerca scientifica ha bisogno di molti ingredienti: cervelli, idee, spirito di iniziativa e, non ultime, risorse. Quello di cui c’è bisogno, in altre parole, è un grosso impegno finalizzato ad implementare la ricerca sulla sindrome di Down, in generale abbastanza penalizzata nel nostro paese, sensibilizzando l’opinione pubblica su un problema che può e deve essere affrontato per ottenere risultati sempre migliori. E’ necessario che si attivi una grande mobilitazione, utilizzando al meglio i mass media, così come è stato fatto con successo per altri grandi problemi della medicina.
La sindrome di Down è vista ancora oggi come un evento che, una volta accaduto, deve essere “serenamente” accettato “perché si può far poco”. Dobbiamo invece tutti augurarci che, in un futuro non troppo remoto, le parole “Il suo bambino ha la sindrome di Down” non facciano più una così grande differenza.
Gina De Bonis e Alvara Sorrentino
Dottorande di Ricerca in Biochimica Cellulare impegnate in una tesi sperimentale sulla sindrome di Down Seconda Università degli Studi Napoli, Dip. “F. Cedrangolo”.
(F.Fimiani*, A.Iovine*, R.Carelli*, M.Pansini*, G.Sebastio**, A.Magli**)
*Department of Opthalmology, University of Neaples “Federico II”
**Department of Pediatry, University of Neaples “Federico II”
Introduzione
La sindrome di Down è una delle anomalie cromosomiche caratterizzate dal punto di vista oculare, da numerose alterazioni, quali anormalità delle palpebre, cornea, iride, cristallino, retina, disco ottico, ambliopia, strabismo, nistagmo. (1)
I principali studi inerenti le anomalie oculari presenti in pazienti con sindrome di Down dimostrano la variabilità delle prevalenza a seconda della popolazione studiata (2-3-4). Il nostro studio descrive le anomalie oculari presenti nei pazienti italiani affetti dalla sindrome di Down.
Materiali e metodi
Sono giunti alla nostra osservazione presso l’ ambulatorio di Oftalmologia Pediatrica e Genetica Oculare del Dipartimento di Scienze Oftalmologiche del Policlinico Universitario degli studi di Napoli Federico II, 157 pazienti di età compresa tra 1 mese e 18 anni, provenienti dalla clinica pediatrica nell’ambito del “Progetto Down” concordato con l’ “Associazione Sindrome di Down” della Regione Campania.
Dei 157 pazienti, 70 maschi e 87 femmine, 81 avevano età compresa tra 0-3 anni, 26 tra 4-6 anni, 16 tra 7-9 anni e 34 tra 9-18 anni. L’età media di questi pazienti era di 5,28 anni, mentre l’età media delle mamme al momento del concepimento era di 31,8 anni. Di questi pazienti 61 (39%) erano nati pretermine. L’anamnesi familiare evidenziava familiarità per sindrome di Down in 3 pazienti (1,9%). I pazienti, precedentemente sottoposti a esame del cariotipo, presentavano le seguenti diagnosi: trisomia 21 libera 153 pz (97,4%); traslocazione 14:21 1pz (0,6%); mosaicismo 2pz (1,2%).
Tutti i pazienti sono stati sottoposti a un esame oftalmologico: dopo l’ispezione del bulbo e dell’ orbita nel suo insieme, è stato effettuato l’esame della motilità oculo-palpebrale, in cui si è valutato la motilità oculare intrinseca ed estrinseca, la presenza di eventuali incomitanze orizzontali e verticali, le escursioni, la fissazione dello sguardo, anomalie della posizione del capo, la conformazione palpebrale, l’ampiezza della rima palpebrale.
È stata misurata l’acuità visiva mediante ottotipo con E di Albini nei pazienti collaboranti di età superiore a 3 anni. Nei pazienti non collaboranti lo studio dell’ acuità visiva è stato condotto con il metodo comportamentale della Direzione Preferenziale di Sguardo a scelta forzata (FPL) nella variante tecnica nota come Acuity Card Procedure (ACP) (5). Successivamente si è praticato l’esame biomicroscopico mediante lampada a fessura per la valutazione del margine palpebrale, della congiuntiva, delle anomalie della cornea, dell’iride e del cristallino.
A tutti i pazienti, previa istillazione di ciclopentolato 1% (3 gocce in entrambi gli occhi ogni 15 minuti), è stato effettuato l’esame schiascopico in cicloplegia.
Sono stati classificati come emmetropi i pazienti con vizi di rifrazione compresi tra -0,50 D (diottrie) e +0,75 D; miopi i pazienti con valori inferiori a -0,50 D ed ipermetropi i pazienti con valori superiori a +0,75 D; astigmatici i pazienti con valori superiori a 0,75 D di cilindro, inoltre sono stati considerati anisometropi i pazienti con una differenza tra i due occhi superiore a 1 D.
Ad alcuni pazienti che presentavano sospetto di glaucoma è stata praticata la tonometria.
Infine tutti i pazienti sono stati sottoposti all’esame oftalmoscopico mediante oftalmoscopio indiretto al fine di esaminare la retina, la coroide e il disco ottico, la macula.
Risultati
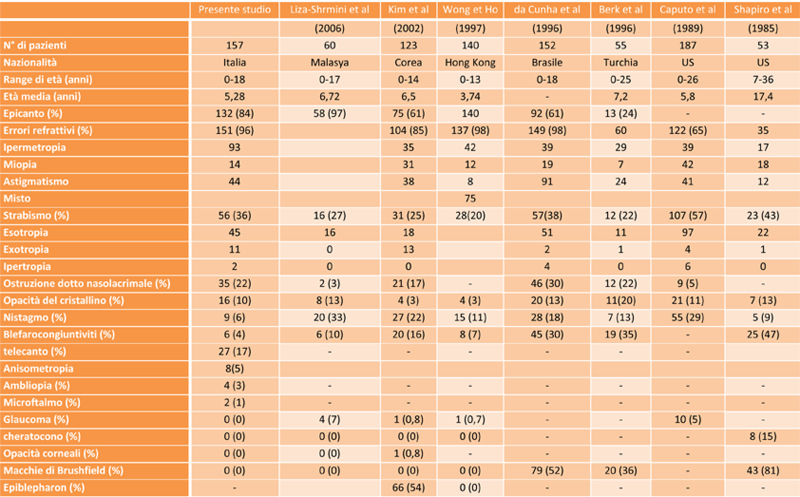
L’incidenza complessiva delle anomalie oculari nei nostri pazienti è del 100% (157/157). I risultati nel dettaglio sono riportati nella tabella 1.
Le anomalie più frequenti sono state i difetti di refrazione, presenti in 151 pazienti (96%), in particolare l’ipermetropia è stata riscontrata in 93 pazienti (59%), l’astigmatismo in 44 pazienti (28%), di cui 27 pazienti presentavano astigmatismo ipermetropico e 14 pazienti presentavano astigmatismo miopico, la miopia in 14 pazienti (9%). L’ epicanto è stato riscontrato in 132 pazienti, (84%).
L’esame della motilità oculare estrinseca ha evidenziato strabismo in 56 pazienti (36%), di cui 45 esotropia e 11 exotropia: sono stati evidenziati 2 pazienti con ipertropia associata a esotropia, un paziente con variazione a V associata a exotropia, e tre pazienti con IOI, di cui una associata ad exotropia, e una associata a esotropia.. Il nistagmo è stato osservato in 9 pazienti (6%). L’ostruzione delle vie lacrimali 35 pazienti (22%) è stata diagnosticata sulla base di una anamnesi positiva per epifora e presenza di rigurgito alla digitopressione in regione del sacco lacrimale.
L’opacità del cristallino è stata riscontrata in 16 pazienti, (10%), blefarocongiuntiviti in 6 pazienti, (4%). Inoltre è stato diagnosticato telecanto (27 pazienti), ambliopia (4 pazienti), ipertelorismo (3 pazienti), microftalmo (2 pazienti), microcornea (1 paziente), calazio della palpebra superiore (1 paziente).
L’esame oftalmoscopico ha evidenziato le seguenti anomalie: corioretinosi miopica (4 pazienti), corioretinosi miopica con stafiloma OS>OD (1 paziente), lieve pallore temporale del nervo ottico (1 paziente), tortuosità vasali (1 paziente), distrofia pigmentaria al polo posteriore tipo degenerazione tapetoretinica (1 paziente), aspetto distrofico OS>OD (1 paziente), lieve emorragia preretinica soprapapillare (1 paziente), tilted disc (1 paziente).
Discussione
Questo studio mostra le differenze tra le varie patologie oculari riscontrate nella sindrome di Down, in base alla razza e all’età.
L’ incidenza complessiva delle anomalie oculari nei bambini italiani affetti dalla sindrome di Down è del 100% (tab.4).
Il nostro studio ha messo in risalto come più frequente anomalia oculare presente nei bambini italiani con Sindrome di Down gli errori di refrazione (96%), un valore simile agli studi di Hong Kong (98%) (6) e del Brasile (98%) (7).
In particolare però i nostri pazienti presentavano una maggiore incidenza di ipermetropia ( 59%), un risultato superiore agli altri studi di cui il valore più alto era rappresentato dallo studio eseguito in Turchia (48%) (3) mentre gli altri studi in letteratura presentavano una incidenza di gran lunga inferiore (1-2-4-6-7) (tab.4).
Questo risultato può essere in parte spiegato per l’ età media dei nostri pazienti che era di 5,28 anni lievemente inferiore agli altri studi eccetto quello eseguito in Cina 3,74 anni (6). La miopia era presente nel (9%), risultato simile allo studio di Wong e Ho (8%), e allo studio di Berk (12%), inferiore rispetto agli altri studi, anche questo risultato spiegabile in parte per l’età media dei nostri pazienti (tab.4).
L’astigmatismo (28%) è simile allo studio della Cina (30%) (6), un risultato accettabile che rientra nei valori riportati in altri studi che vanno dal 22% al 60% (tab.4). L’ incidenza di strabismo era del (36%), risultato paragonabile allo studio di Wong e Ho (38%), in particolare sul numero totale di strabismo l’ exotropia era presente nel 20% dei pazienti strabici un risultato che a parte lo studio eseguito in Korea (42%), è paragonabile allo studio di Hiles (19%)(10) ed è superiore di gran lunga agli altri studi (tab.3).
Il nistagmo presentava un’ incidenza del (6%), risultato paragonabile allo studio di Eissler e Longenecker (4%) (8), ma inferiore rispetto agli altri studi riportati nella tabella 4. La cataratta nei bambini con sindrome di Down insorge soprattutto tra i 12-15 anni e quasi mai prima dei 6 anni (9) da sottolineare tuttavia che l’ incidenza delle opacità lenticolari nel nostro studio è stata del (10%), simile allo studio di Caputo (11%), di queste opacità riscontrate, 13 erano di tipo bilaterale e 3 erano di tipo monolaterale per un totale di 29 occhi. I tipi di opacità osservati sono descritti nella tabella 5. La cataratta è risultata essere sicuramente congenita in due pazienti che sono stati operati.
L’ostruzione delle vie lacrimali (22%) è simile allo studio della Turchia (22%) in accordo con i risultati riportati in altri studi che in genere varia tra 5% e 30%. Le blefarocongiuntiviti sono state riscontrate nel (4%), valore paragonabile allo studio di Wong e Ho (7%), ma inferiore agli altri studi riportati nella (tab.4).
L’incidenza di cheratocono nella sindrome di Down varia tra 0% e 30% non sorprende che non c’erano casi nei nostri pazienti. L’epicanto era presente nei nostri pazienti (84%), risultato che rientra nel range di altri studi che varia dal 9% al 100% (10,12,14) (tab.4). Assenza di glaucoma e cheratocono.
Conclusioni
I pazienti italiani con sindrome di Down rispetto agli altri studi presentavano nell’ambito dei vizi di refrazione una maggiore incidenza di ipermetropia ed erano meno miopi. Presentavano meno nistagmo e blefarocongiuntiviti, mentre lo strabismo è ben presente in particolare da notare la percentuale di exotropia (20%) sul totale degli strabici. Da considerare anche la presenza di ostruzione delle vie lacrimali nel (22%). Assenza di cheratocono e glaucoma.
Bibliografia:
1) Shapiro MB, France TD. The ocular features of Down’s sindrome. American Journal Ophthalmology 1985; 99: 659-663
2) Caputo AR, Wagner RS, Reynolds DR, Guo S, Goel AK. Down Syndrome. Clinical review of ocular features. Clinical Pediatrics 1989; 28: 355-358
3) Berk AT, Saatci AO, Ercal MD, Tunc M, Ergin M. Ocular findings in 55 patients with Down’s syndrome. Ophthalmic Genetics 1996; 17: 15-19
4) Kim JH, Hwang JM, Kim HJ, Yu YS. Characteristic ocular findings in Asian children with Down syndrome. Eye 2002; 16: 710-714
5) Amabile S, Bertoni T, Magli A. Studio dell’acuità visiva binoculare su una popolazione pediatrica di soggetti con Syndrome di Down. Bollettino di oculistica 1996 Anno 75- Gennaio-Febbraio- N.1 251-257
6) Wong V, Ho D. Ocular abnormalities in Down Syndrome: An Analysis of 140 Chinese Children. Pediatr Neurol 1997; 16: 311-314
7) da Cunha RP, Moreira JB. Ocular findings in Down’s syndrome. American Journal Ophthalmology 1996; 122: 236-244
8) Eissler R, Longenecker LP. The common eye findings in mongolism. American Journal Ophthalmology 1962; 54: 398-406
9) Igersheimer J. The relationships of lenticular changes in mongolism. Trans Am Ophthalmol Soc 1951; 45: 595-624
10) Solomons G, Zellwegger H, Jahnke PG, Optiz E. Four common eye signs in mongolism. Am J Dis Child 1965; 110: 46-50
11) Hiles DA, Hoyme SH, McFarlane F. Down’s syndrome and strabismus. Am Orthopt J 1974; 24: 63-68
12) Liza-Sharmini AT, Azlan ZN, Zilfalil BA. Ocular findings in Malaysian children with Down sindrome. Singapore Med J 2006; 47(1):14-19
13) Lowe RF. The eyes in mongolism. British Journal Ophthalmology 1949; 33: 131-154
14) Roizen NJ, Mets MB, Blondis TA. Ophthalmic disorders in children with Down syndrome. Dev Med Child Neurol 1994; 36: 594-600
La giornata inaugurale della serie di incontri organizzati per il ventennale dell’A.S.D., tenutasi presso 1’Antisala dei Baroni del Maschio Angioino, ha visto l’intervento, fra gli altri, del Prof. Andrea Ballabio chiamato come professore di Genetica Umana presso 1’Università Federico II di Napoli e Direttore del TELETHON Institute of Genetics and Medicine di Napoli.
Il Prof. Ballabio ha illustrato i risultati della ricerca effettuata da vari gruppi internazionali tra i quali anche quello da lui diretto. Tali risultati, pubblicati su “Nature”, una delle più prestigiose riviste scientifiche internazionali, hanno avuto l’obiettivo di contribuire alla comprensione delle conseguenze della trisomia 21.
È risultato, infatti, sempre difficile ai genetisti capire perché tre dosi di geni non alterati, contenuti in un cromosoma, invece delle due dosi normali comportino tante complesse anomalie a carico di vari sistemi, organi ed apparati, come i sistemi nervoso, immunitario, endocrinologico, il cuore e l’apparato digerente.
In tale ricerca si è studiato il “livello di funzionamento” (acceso/spento) nei diversi organi di 200 geni di topo corrispondenti a quelli contenuti nel cromosoma 21 umano. Tale ricerca ha costruito così un vero e proprio atlante di come vengono espressi i geni omologhi a quelli presenti nel cromosoma 21 umano, in particolare negli organi più colpiti nella sindrome di Down, quali il sistema nervoso centrale, il cuore, l’apparato gastrointestinale e gli arti.
Queste ricerche rappresentano il punto di partenza per capire la funzione e la regolazione dei geni contenuti nel cromosoma 21 e comprendere perché lo sbilanciamento in eccesso di questi geni causi così importanti alterazioni nella sintomatologia sindromica.
Prof. G. Andria
Si riportano le dichiarazioni fatte dal Prof. Ballabio durante la sua esposizione della ricerca fatta sulla trisomia 21
“Mi fa particolarmente piacere questa coincidenza perché proprio in questi giorni noi stiamo per festeggiare, come Istituto, un’importante scoperta scientifica.
Sono nato a Napoli, mi sono laureato a Napoli e specializzato in Pediatria, nell’ambito della Pediatria mi sono appassionato di malattie genetiche ed ereditarie.
Sono andato all’estero per specializzarmi ulteriormente ed ho imparato anche ad organizzare la ricerca. Mi è stata data la possibilità da Telethon di poter rientrare a Napoli e continuare a fare ricerca ad alto livello.
L’Associazione Telethon si occupa di ricerca, del sociale e della comunicazione…, in realtà la ricerca è molto importante perché può risolvere i problemi ma i tempi della Ricerca sono più lunghi di quelli del Sociale e della Comunicazione e questo è il motivo perché purtroppo i fondi spesso vengono tagliati, perché i risultati tardano ad arrivare. Per fortuna nella Regione Campania non stiamo molto male. Due anni fa noi di Telethon ci siamo trasferiti a Napoli. L’intero Istituto di ricerca si è trasferito insieme ai ricercatori milanesi, diventando un’eccezione positiva rispetto alla migrazione verso l’estero che di solito avviene. Veniamo adesso alla ricerca.
La sindrome di Down è una sindrome complessa e la complessità è a vari livelli. Un primo livello è dato dal fatto che la sindrome coinvolge organi e tessuti diversi e sintomatologicamente apparentemente non correlati. Quindi ci sono tanti livelli diversi.
Un secondo livello, che la rende più difficile da studiare è che si tratta di una condizione genetica, ma in questo caso non è a carico di un solo gene, come in altre situazioni, bensì coinvolge vari geni e poiché il cromosoma 21 è presente in eccesso, ci sono più copie di tanti geni ed i sintomi devono esser ricondotti ai geni che determinano queste risposte. Generalmente siamo abituati a studiare la funzione di un singolo gene.
Il terzo livello di complessità è che abitualmente, nelle alterazioni genetiche, si studiano i geni “mancanti”, che non funzionano e invece nella trisomia bisogna studiare i geni “in eccesso” cioè che funzionano troppo e dobbiamo capire cosa succede con una funzionalità eccessiva.
Oggi siamo ad una svolta grazie ad un grosso progetto Internazionale, il “Progetto Genoma” il cui fine è quello di decifrare l’intero patrimonio genetico dell’Uomo e quindi comprendere e decodificare la successione di lettere del nostro patrimonio genetico. Il nostro patrimonio genetico è paragonabile ad una Enciclopedia, all’interno della quale c’è scritto come nasciamo, cresciamo, ci riproduciamo, ecc., Questa si suddivide in 46 Volumi (che sarebbero i cromosomi). Nella nostra enciclopedia però i volumi sono presenti in coppie, 23 coppie di Volumi (i 46 cromosomi) e nella sindrome di Down, il volume N° 21 è presente in tre copie. Cosa c’è scritto nel volume N° 21? All’interno del volume ci sono tanti geni cioè i Capitoli, e ci sono circa 200 geni. I geni contengono un messaggio preciso (così come i capitoli) per esempio come sintetizzare una proteina per l’emoglobina, o per la funzione cardiaca, ecc.
Adesso questo patrimonio genetico è stato decifrato ma ad oggi dobbiamo sapere a che cosa servono i geni presenti nel cromosoma 21, a che cosa servono per tutti noi, e quindi anche comprendere cosa succede quando lavorano in eccesso e perché. Il nuovo progetto al quale stiamo lavorando da circa due anni, mira a decifrare tutti i geni presenti nel cromosoma 21 ed in particolare studia l’aspetto della distribuzione della funzione nei tessuti del cromosoma 21, cioè in quali organi operano. I geni sono presenti in tutte le cellule del nostro corpo però esercitano la loro funzione a seconda se sono “accesi” o “spenti”, ad esempio alcuni geni servono per la funzionalità del cervello, altri per il fegato, per il cuore, ecc., cioè non tutti esercitano la stessa funzione, non tutti sono accesi o spenti negli stessi tessuti.
Altra complessità è che alcune anomalie della sindrome sono congenite ed altre possono svilupparsi dopo, durante la crescita (processo ossidativo ed invecchiamento precoce), per cui i geni sono studiati durante lo sviluppo, in particolare questo è stato possibile nello studio del Topo, (n.d.r.: il cui corredo genetico è simile a quello del cromosoma 21 umano) ed abbiamo tracciato un Atlante, un vero e proprio atlante anatomico, che è stato pubblicato su “Nature” in cui si vedono tutti i tessuti e le strutture dei tessuti, si vede in quale tessuto è “acceso” o “spento”. Nel cromosoma 21 ci sono circa 200 geni ed è stato notato che circa una dozzina sono espressi, cioè esercitano la loro funzione sul cuore e sullo sviluppo del cuore, altri nel tessuto valvolare e possono essere responsabili delle anomalie delle valvole cardiache, altri geni sono presenti solo ed esclusivamente nelle bozze degli arti, durante lo sviluppo embrionale e dopo non funzionano più, alcuni servono per lo sviluppo dell’intestino, e molti per lo sviluppo del sistema nervoso centrale e in parte anche del cervello, pensiamo che lì risieda la causa del possibile ritardo mentale.
In questa mappa, bisognerà focalizzare i geni che sono coinvolti in queste patologie ed in particolare negli animali noi possiamo provocare l’eccesso della produzione dei geni e quindi vedere come “contrastare” quest’eccesso di funzione e correggere l’eventuale difetto.
Un’altra cosa che si deve capire è perché esiste una diversità dei sintomi tra persona e persona, per poter predisporre una eventuale linea d’intervento comune. La speranza è che si possa arrivare a veri e propri interventi che abbiano una ricaduta positiva sulle persone con sindrome di Down per una migliore qualità della vita. Prima di concludere vorrei citare alcuni dei miei colleghi, in particolare Valeria Marigo e Sandro Banfi che sono ricercatori informatici, gran parte del lavoro è stato fatto grazie agli ausili informatici, così un grazie ai finanziamenti Telethon che rendono possibile il procedere della ricerca…”
Prof. Andrea Ballabio
